Overview
Introduction
Peptide-protein interactions play a critical role in the protein-protein interaction network with significant involvement in signal transduction and regulation. Many of these interactions are promising candidates for providing new leads for drug targets. Thus the development of computational tools for accurate and efficient modeling of these interaction is of high priority.
Peptides often lack a distinct fold in their unbound state, and go through simultaneous binding and folding upon encountering their target protein receptor. In many cases no knowledge regarding the peptide binding site and/or the peptide backbone conformation is available. Structure-based modeling of these interactions is very challenging. A high-resolution global peptide-protein docking protocol is required to address such challenging modeling problems.
Significant progress has been made recently with the development of many peptide-protein docking algorithms. These significant recent advances in global peptide docking notwithstanding, present approaches are still limited in their modeling quality and general applicability, and there is ample room for improvements that would enable the detailed high-resolution study of more peptide-protein interactions with higher accuracy. We have developed PIPER-FlexPepDock, a high resolution global peptide-protein docking protocol for more accurate and efficient high-resolution modeling of peptide-protein interactions.
Protocol Outline
PIPER-FlexPepDock uses a divide and conquer approach to reduce the complexity associated with modeling highly flexible peptide on large receptor surfaces. By streamlining the Rosetta fragment picker for accurate peptide fragment ensemble generation, the PIPER docking algorithm for exhaustive fragment -receptor rigid-body docking and Rosetta FlexPepDock for flexible full-atom refinement of PIPER docked models, it successfully addresses the challenge of accurate and efficient global peptide-protein docking at high-resolution with remarkable accuracy.

Figure 1. Overview of the PIPER-FlexPepDock peptide docking protocol. Example shown: PDZ domain-peptide interaction, with PDB IDs of receptor structure 1MFG (bound) and 2H3L (free)]. For a given receptor structure and peptide sequence, the divide and conquer strategy involves first the description of the peptide as an ensemble of fragments (A), their fast and exhaustive rigid body docking (using PIPER) onto the receptor (binding site region is shaded salmon) (B), and subsequent high-resolution refinement (using Rosetta FlexPepDock; the top 5000 models are included in the plot.) (C), followed by clustering and selection of top ranking representatives.
Performance & Benchmarking
PIPER-FlexPepDock has been thoroughly benchmarked against a set of 27 non-redudant solved peptide-protein complexes taken from the PeptiDB data set. For the bound runs (receptor was taken from the solved complex structure) a near-native peptide conformation (L-RMSD= 2.0Å) was found among the top 10 ranked clusters for 19 out of 27 complexes (success rate=70%). For the unbound runs where the free receptor structure was provided as starting point 11 complexes modeled successfully (success rate=41%) using the same evaluation criteria. Importantly however, when including also receptor flexibility during the refinement stage, these results improved with 14 complexes being modeled accurate among thw top 10 ranking cluster representatives (success rate=52%).
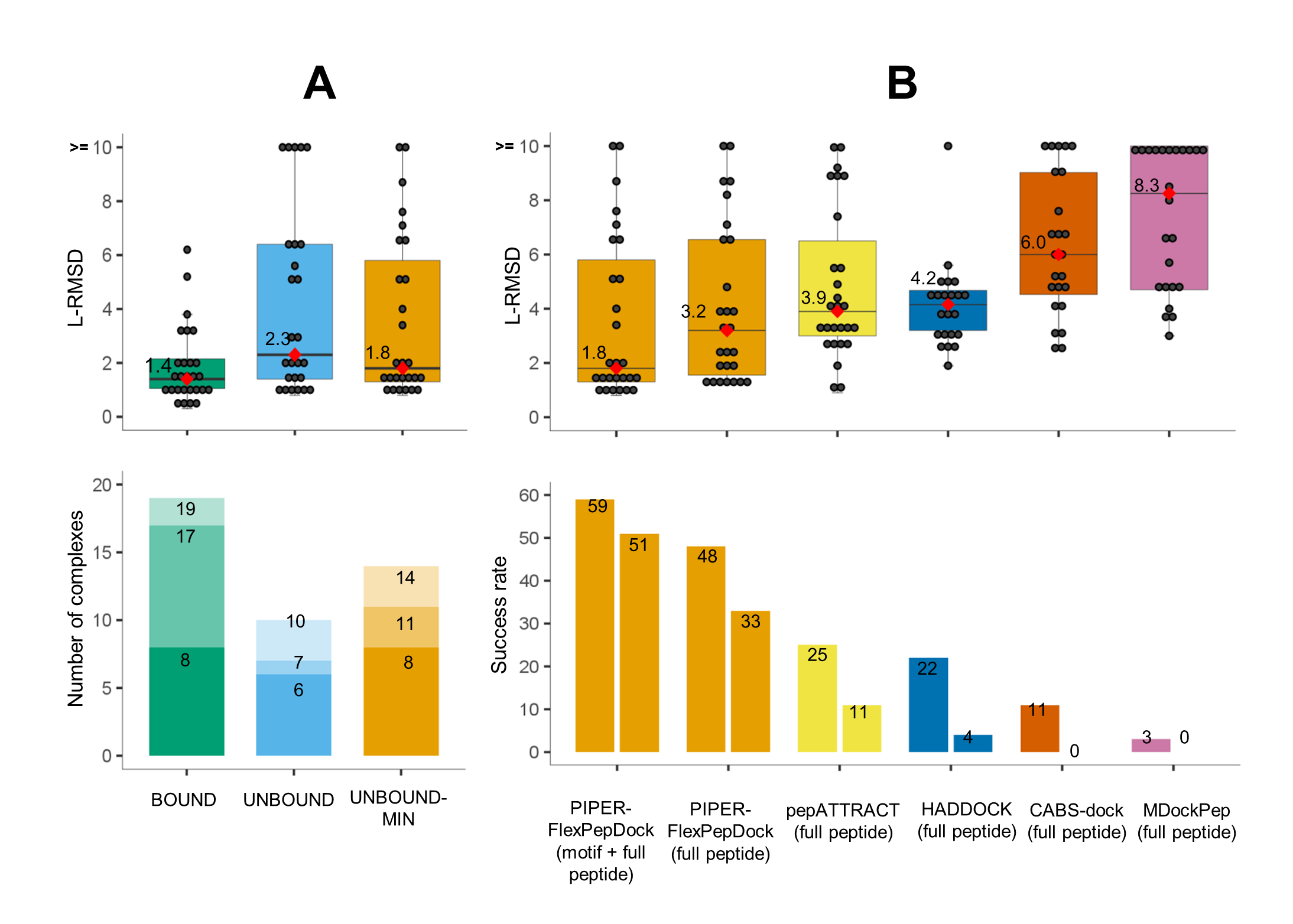
Figure 2. PIPER-FlexPepDock peptide docking performance. (A) Overall performance on a non-redundant set of 27 peptide-protein complexes. Top: Distribution of best model L-RMSDs (among top 10 ranking clusters) for runs using the bound receptor structure (BOUND) and the free receptor structure (UNBOUND & UNBOUND-MIN), the latter including also receptor flexibility in the final refinement step (Only the motif region was modeled for the 12 complexes with known motif). The median values are shown as red diamonds and printed alongside. Bottom: Distribution of the ranks of the first near-native cluster (defined as L-RMSD <=2.0A) shown using different shades (for corresponding results among the top1, top3 and top10 ranked predictions). (B) Comparison to performance by other algorithms. Top: Box plots of best L-RMSDs among top 10 ranking clusters, including results for the motif part where the motif is known (as in A), as well as for the full peptide, for comparison. Bottom: Performance is shown for different cutoffs (2.0 and 3.0 L-RMSD, left and right boxes).
References
Please cite the following when referring to results from our server:
- Alam, Goldstein, Xia, Porter, Kozakov, Schueler-Furman. High-resolution global peptide-protein docking using fragments-based PIPER-FlexPepDock. (2017) PLoS CB 13:e1005905
- Porter KA, Xia B, Beglov D, Bohnuud T, Alam N, Schueler-Furman O, Kozakov D. ClusPro PeptiDock: Efficient global docking of peptide recognition motifs using FFT. (2017) Bioinformatics btx216
- Raveh B, London N, Schueler-Furman O. Sub-angstrom modeling of complexes between flexible peptides and globular proteins. (2010) Proteins 78:2029-40
- Kozakov D, Brenke R, Comeau SR, Vajda S. PIPER: An FFT-based protein docking program with pairwise potentials. (2006) Proteins : Structure, Function, and Bioinformatics 65:392–406